Theoretical Investigation of New Possible Fatty Hydrazides Corrosion Inhibitors Via Density Functional Theory
DOI:
https://doi.org/10.37934/armne.15.1.1421Keywords:
Corrosion inhibitors, fatty hydrazides derivatives, density functional theory, inhibition efficiencyAbstract
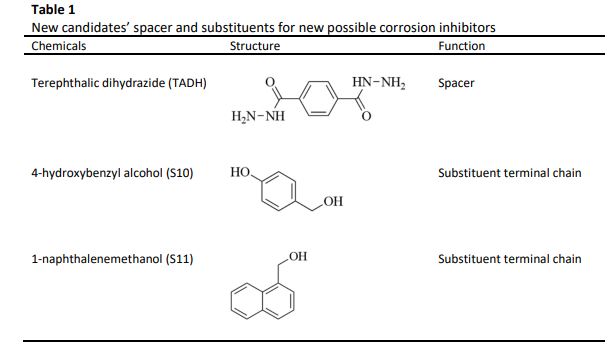
In this study, two new possible corrosion inhibitors derived from fatty hydrazides were investigated via the density functional theory (DFT) to measure the quantum chemical calculation of these new molecules and analyse their corrosion inhibition ability. The quantum calculation parameters included the calculation for the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO) and the energy gap between HOMO and LUMO. Other chemical descriptors were also measured, including the global hardness, electronegativity, and the fraction of transferred electrons. The results revealed that both new possible corrosion inhibitors, TADHS11 and TADHS12, showed excellent chemical reactivity and inhibition efficiency based on the calculation of electronic properties, with band gap energy of 4.35 eV for TADHS10 and 4.18 eV for TADHS11. The other chemical descriptor also revealed similar observations, which agree that these two compounds exhibit excellent corrosion inhibitor properties. Overall, all data were consistent with previously reported findings, proving the ability of these new possible fatty hydrazide derivatives as effective corrosion inhibitors.